
有機半導体の移動度をシミュレーションで予測:材料開発の時間や労力を軽減
筑波大学や東京大学らの共同研究グループは、分子の化学構造式と粉末X線回折パターンを用いて、有機半導体の移動度を予測するシミュレーション法を開発した。高性能な有機半導体の材料開発に要していた作業時間と労力を大幅に削減することが可能となる。
化学構造式と粉末X線回折データを活用
筑波大学や東京大学らの共同研究グループは2020年2月、分子の化学構造式と粉末X線回折パターンを用いて、有機半導体の移動度を予測するシミュレーション法を開発したと発表した。高性能な有機半導体の材料探索にはこれまで、単結晶の作製などが必要であったが、開発した手法を用いると材料開発の作業時間と労力を大幅に削減することができるという。
今回の研究成果は、筑波大学数理物質系の石井宏幸助教と小林伸彦教授、コンフレックスの小畑繁昭研究員、豊橋技術科学大学情報・知能工学系の後藤仁志准教授、東京大学大学院新領域創成科学研究科の竹谷純一教授と岡本敏宏准教授、渡邉峻一郎特任准教授らによるものである。
有機半導体は低温プロセスで印刷が可能なことから、次世代電子材料として注目されている。ただ、高性能な半導体デバイスを実現するには、移動度の高い有機半導体材料を開発していく必要がある。
ところがこれまでの材料開発では、新たな有機半導体分子の合成から単結晶の作製、トランジスタの作製および、試作したチップの移動度評価など、多くの作業を繰り返し行う必要があるため、開発効率が課題となっていた。
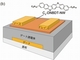
共同研究グループは今回、分子の化学構造式と粉末X線回折パターンから、単結晶構造と移動度などの材料特性を短期間で予測するシミュレーション法を開発した。従来方式とは異なり、単結晶構造の測定データを用いずに、高い精度で移動度を予測することができるのが特長だ。

具体的には、筑波大学の石井氏と小林氏らが、予測構造に対する移動度の大きさと温度依存性を速やかに予測する「大規模量子伝導シミュレーション法」を開発。コンフレックスの小畑氏と豊橋技術科学大学の後藤氏らが、網羅的な結晶構造探索とエネルギー評価による「結晶構造予測シミュレーション法」を開発した。
今回はこれらのシミュレーション手法に新しい評価法を導入した。それは、予測の精度向上と時間短縮を可能にするもので、大きな単結晶よりも簡便に得られる粉末結晶のX線回折パターンを利用した。
開発したシミュレーション手法を、東京大学の岡本氏や竹谷氏らが開発した高性能半導体分子「C10-DNBDT」に適用した。この結果、渡邉氏や竹谷氏らが測定によって明らかにした結晶構造やトランジスタ移動度を、高い精度で再現できることが分かった。

今回用いた大規模量子伝導シミュレーション法は、温度差により発電する熱電物性の計算にも拡張できる。このため、有機半導体の移動度に加え、「熱電物性や熱伝導などの機能予測にも活用することが可能」と研究グループはみている。
 遮断周波数が38MHzの有機トランジスタを開発
遮断周波数が38MHzの有機トランジスタを開発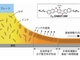 有機半導体の分子形状を物理吸着で一斉に制御
有機半導体の分子形状を物理吸着で一斉に制御 東大、シールのように貼れる有機半導体膜を開発
東大、シールのように貼れる有機半導体膜を開発 東大ら、簡便な印刷法で有機半導体ウエハー作製
東大ら、簡便な印刷法で有機半導体ウエハー作製 高移動度有機半導体を実現する結晶構造制御法開発
高移動度有機半導体を実現する結晶構造制御法開発 有機半導体の微結晶試料から物質固有の移動度予測
有機半導体の微結晶試料から物質固有の移動度予測Copyright © ITmedia, Inc. All Rights Reserved.
Special Contents
- PR -
- [EE Times Japan驍ア�ィ鬮ョ�スホ咯 EE Times Japan��窮DN Japan 驍ィ�ア陷キ逎ッ蟠戊氛蜊�イソ�ス螢シ蛻磯ィセ貅倪�郢ァ荵敖蠕後℃郢晢スゥ郢ァ�ケ陜難スコ隴夲スソ邵イ蝓シ蟷暮具スコ邵イツ隴鯉ス・隴幢スャ郢ァ繧�ソ�郢ァ轣假スシ蜍滂スス�ケ邵コ�ォ遯カ陬慊�ス 鬮ョ�サ陝�鴻豐ソ2025陝キ�エ4隴帑コ・謐キ
- 鬮「迢怜験郢ァ蝣、�ー�。驍擾ソス陋ケ謔カ��驕カ�カ闔�迚咎エィ郢ァ雋橸スシ�キ陋ケ謔カ笘�ケァ荵敖竏ォ�ャ�ャ4闕ウ邏具スサ�」雎主ョ育舞USB郢晢ソス繝ー郢ァ�、郢ァ�ケ郢ァ�ウ郢晢スウ郢晏現ホ溽ケ晢スシ郢晢スゥ郢晢スシ邵コ�ョ陞ウ貅キ魘ィ
- 陷企宦�ー諠ケ�ス隰趣スク蟠取�髫暦ス」隴ォ闊鯉ソス隴�スー陟�ソス諷咲クイ竏夲ソス郢晢スュ郢ァ�サ郢ァ�ケ鬮「迢怜験郢ァ雋槫�鬨セ貅倪�郢ァ蛹コ�ャ�。闕ウ邏具スサ�」邵コ�ョ3D X驍ア螟撰ス。蜍滂スセ�ョ鬮�。邵コ�ョ陞ウ貅キ魘ィ
- 邵イ謔溷ア楢ャ壼・縺慕ケ晢スウ郢晏現ホ帷ケァ�ケ郢晏現繝ィ郢晢ス「郢ァ�ー郢晢スゥ郢晁シ斐≦邵イ髦ェ�帝$逧ョ�ゥ�カ陞ウ�、郢晢スャ郢晏生ホ晉クコ�ァ陞ウ貅ス讓溽クコ蜷カ�玖ュ�スケ雎戊シ披�邵コ�ッ�ス�ス
- [EE Times Japan驍ア�ィ鬮ョ�スホ咯 陷企宦�ー諠ケ�ス轣倩�驕会スセ 隶鯉スュ驍オ�セ邵コ�セ邵コ�ィ郢ァ竏堋�ス2025陝キ�エ3隴帛沺謔�圷�ャ3陜怜ク帶ソ�隴帶コ伉�ス

Special Contents 2
- PR -

記事ランキング
- 半導体メーカーの「悲喜こもごも」 絶好調のTSMC、人員削減のST
- IntelがAltera売却へ、株式51%を米投資ファンドに
- 2024年の半導体市場は21%成長 NVIDIAが初の首位に
- 世界半導体市場が10カ月連続で17%以上成長 2月として過去最高に
- チップに「水路」を作り冷却液を流し込む 高効率に放熱
- ミネベアミツミが芝浦電子買収へ 「8本槍」戦略強化に向け
- AIのデータ転送問題解決に王手、シリコンフォトニクス新興企業
- トランプ政権の「アメとムチ」 Intelは補助金を受け取れるのか
- 2035年のウエハー需要を予測する ~半導体も「VUCA時代」に
- 半導体デバイスの発熱を制御するメカニズムを発見
Special Site
- PR -

![]() ITmediaはアイティメディア株式会社の登録商標です。
ITmediaはアイティメディア株式会社の登録商標です。